Signac: Integration and clustering of chromap quantifications
Last updated: 2022-09-14
Checks: 7 0
Knit directory: chromap_vs_cellranger_scATAC_exploration_10x/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220912) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 10fdcb0. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
working directory clean
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ChromapSignacIntegration.Rmd) and HTML (docs/ChromapSignacIntegration.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 10fdcb0 | jeremymsimon | 2022-09-14 | Initial commit |
Load packages and previously saved workspace
library(tidyverse)── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──✔ ggplot2 3.3.6 ✔ purrr 0.3.4
✔ tibble 3.1.8 ✔ dplyr 1.0.9
✔ tidyr 1.2.0 ✔ stringr 1.4.0
✔ readr 2.1.2 ✔ forcats 0.5.1── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(GenomicRanges)Loading required package: stats4Loading required package: BiocGenerics
Attaching package: 'BiocGenerics'The following objects are masked from 'package:dplyr':
combine, intersect, setdiff, unionThe following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which.max, which.minLoading required package: S4Vectors
Attaching package: 'S4Vectors'The following objects are masked from 'package:dplyr':
first, renameThe following object is masked from 'package:tidyr':
expandThe following objects are masked from 'package:base':
expand.grid, I, unnameLoading required package: IRanges
Attaching package: 'IRanges'The following objects are masked from 'package:dplyr':
collapse, desc, sliceThe following object is masked from 'package:purrr':
reduceLoading required package: GenomeInfoDblibrary(Seurat)Attaching SeuratObjectlibrary(Signac)
library(EnsDb.Hsapiens.v86)Loading required package: ensembldbLoading required package: GenomicFeaturesLoading required package: AnnotationDbiLoading required package: BiobaseWelcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'AnnotationDbi'The following object is masked from 'package:dplyr':
selectLoading required package: AnnotationFilter
Attaching package: 'ensembldb'The following object is masked from 'package:dplyr':
filterThe following object is masked from 'package:stats':
filterload("cCRE_hg38_10x_HGMM_PBMC_chromap_fragments_MACS_q01_unionPeaks_merge100_seurat_090222_QC.RData")Merge chromap seurat objects, subset, then split into a list
chromap.combined <- merge(HGMM_10x_seurat,y = PBMC_10x_seurat)Warning: Feature names cannot have underscores ('_'), replacing with dashes
('-')# Subset for features observed in at least 20 cells
filtFeatures <- FindTopFeatures(chromap.combined,min.cutoff=20)
chromap.combined <- subset(chromap.combined, features = VariableFeatures(filtFeatures))
# Split by sample into a list
chromap.list <- SplitObject(chromap.combined, split.by="Sample")Iterate over sample list, subset based on QC metrics, and generate lsi embeddings
for (i in 1:length(chromap.list)) {
chromap.list[[i]] <- subset(x = chromap.list[[i]],
features = VariableFeatures(filtFeatures),
subset = nCount_peaks > 1000 &
nCount_peaks < 100000 &
FRiP > 0.15 &
blacklist_fraction < 0.05 &
nucleosome_signal < 4 &
TSS.enrichment > 2
)
chromap.list[[i]] <- FindTopFeatures(chromap.list[[i]], min.cutoff = 'q0')
chromap.list[[i]] <- RunTFIDF(chromap.list[[i]], assay="peaks")
chromap.list[[i]] <- RunSVD(chromap.list[[i]])
chromap.list[[i]] <- RunUMAP(chromap.list[[i]], reduction = "lsi", dims = 2:30)
}Performing TF-IDF normalizationWarning in RunTFIDF.default(object = GetAssayData(object = object, slot =
"counts"), : Some features contain 0 total countsRunning SVDScaling cell embeddingsWarning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per session12:28:17 UMAP embedding parameters a = 0.9922 b = 1.11212:28:17 Read 5636 rows and found 29 numeric columns12:28:17 Using Annoy for neighbor search, n_neighbors = 3012:28:17 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
12:28:18 Writing NN index file to temp file /tmp/Rtmp4AqXVl/filee43d7377b738
12:28:18 Searching Annoy index using 1 thread, search_k = 3000
12:28:20 Annoy recall = 100%
12:28:28 Commencing smooth kNN distance calibration using 1 thread
12:28:41 Initializing from normalized Laplacian + noise
12:28:41 Commencing optimization for 500 epochs, with 221718 positive edges
12:28:57 Optimization finished
Performing TF-IDF normalizationWarning in RunTFIDF.default(object = GetAssayData(object = object, slot =
"counts"), : Some features contain 0 total countsRunning SVD
Scaling cell embeddings
12:35:09 UMAP embedding parameters a = 0.9922 b = 1.112
12:35:09 Read 11236 rows and found 29 numeric columns
12:35:09 Using Annoy for neighbor search, n_neighbors = 30
12:35:09 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
12:35:13 Writing NN index file to temp file /tmp/Rtmp4AqXVl/filee43d30218077
12:35:13 Searching Annoy index using 1 thread, search_k = 3000
12:35:19 Annoy recall = 100%
12:35:28 Commencing smooth kNN distance calibration using 1 thread
12:35:42 Initializing from normalized Laplacian + noise
12:35:42 Commencing optimization for 200 epochs, with 459300 positive edges
12:35:57 Optimization finished# Subset and process the combined dataset
chromap.combined <- subset(
x = chromap.combined,
features = VariableFeatures(filtFeatures),
subset = nCount_peaks > 1000 &
nCount_peaks < 100000 &
FRiP > 0.15 &
blacklist_fraction < 0.05 &
nucleosome_signal < 4 &
TSS.enrichment > 2
)Get filtered dimensions
table(chromap.combined$Sample)
HGMM PBMC
5636 11236 lapply(chromap.list, dim)$HGMM
[1] 713685 5636
$PBMC
[1] 713685 11236Repeat here on the combined object
chromap.combined <- FindTopFeatures(chromap.combined, min.cutoff = 'q0')
chromap.combined <- RunTFIDF(chromap.combined, assay="peaks")Performing TF-IDF normalizationchromap.combined <- RunSVD(chromap.combined)Running SVDScaling cell embeddingschromap.combined <- RunUMAP(chromap.combined, reduction = "lsi", dims = 2:30)12:45:09 UMAP embedding parameters a = 0.9922 b = 1.11212:45:09 Read 16872 rows and found 29 numeric columns12:45:09 Using Annoy for neighbor search, n_neighbors = 3012:45:09 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
12:45:14 Writing NN index file to temp file /tmp/Rtmp4AqXVl/filee43d381c167
12:45:14 Searching Annoy index using 1 thread, search_k = 3000
12:45:24 Annoy recall = 100%
12:45:34 Commencing smooth kNN distance calibration using 1 thread
12:45:45 Initializing from normalized Laplacian + noise
12:45:47 Commencing optimization for 200 epochs, with 667204 positive edges
12:46:08 Optimization finishedIntegrate all data
# Find integration anchors
integration.anchors <- FindIntegrationAnchors(object.list = chromap.list,
anchor.features = rownames(chromap.combined),
reduction = "rlsi",
dims = 2:30
)Computing within dataset neighborhoodsFinding all pairwise anchorsWarning: No filtering performed if passing to data rather than countsWarning: Feature names cannot have underscores ('_'), replacing with dashes
('-')Projecting new data onto SVDProjecting new data onto SVDFinding neighborhoodsFinding anchors Found 129 anchors# Integrate LSI embeddings
# Arbitrarily reduced k.weight to 50 to get around "number of items to replace is not a multiple of replacement length" error
chromap.integrated <- IntegrateEmbeddings(anchorset = integration.anchors,
reductions = chromap.combined[["lsi"]],
new.reduction.name = "integrated_lsi",
dims.to.integrate = 1:30,
k.weight = 50
)Warning: Feature names cannot have underscores ('_'), replacing with dashes
('-')Merging dataset 1 into 2Extracting anchors for merged samplesFinding integration vectorsFinding integration vector weightsIntegrating dataCreate a new UMAP using the integrated embeddings
chromap.integrated <- RunUMAP(chromap.integrated, reduction = "integrated_lsi", dims = 2:30)12:56:10 UMAP embedding parameters a = 0.9922 b = 1.11212:56:10 Read 16872 rows and found 29 numeric columns12:56:10 Using Annoy for neighbor search, n_neighbors = 3012:56:10 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
12:56:13 Writing NN index file to temp file /tmp/Rtmp4AqXVl/filee43d1a73aff2
12:56:13 Searching Annoy index using 1 thread, search_k = 3000
12:56:19 Annoy recall = 100%
12:56:25 Commencing smooth kNN distance calibration using 1 thread
12:56:33 Initializing from normalized Laplacian + noise
12:56:34 Commencing optimization for 200 epochs, with 671784 positive edges
12:56:46 Optimization finishedDimPlot(chromap.integrated, group.by = "Sample")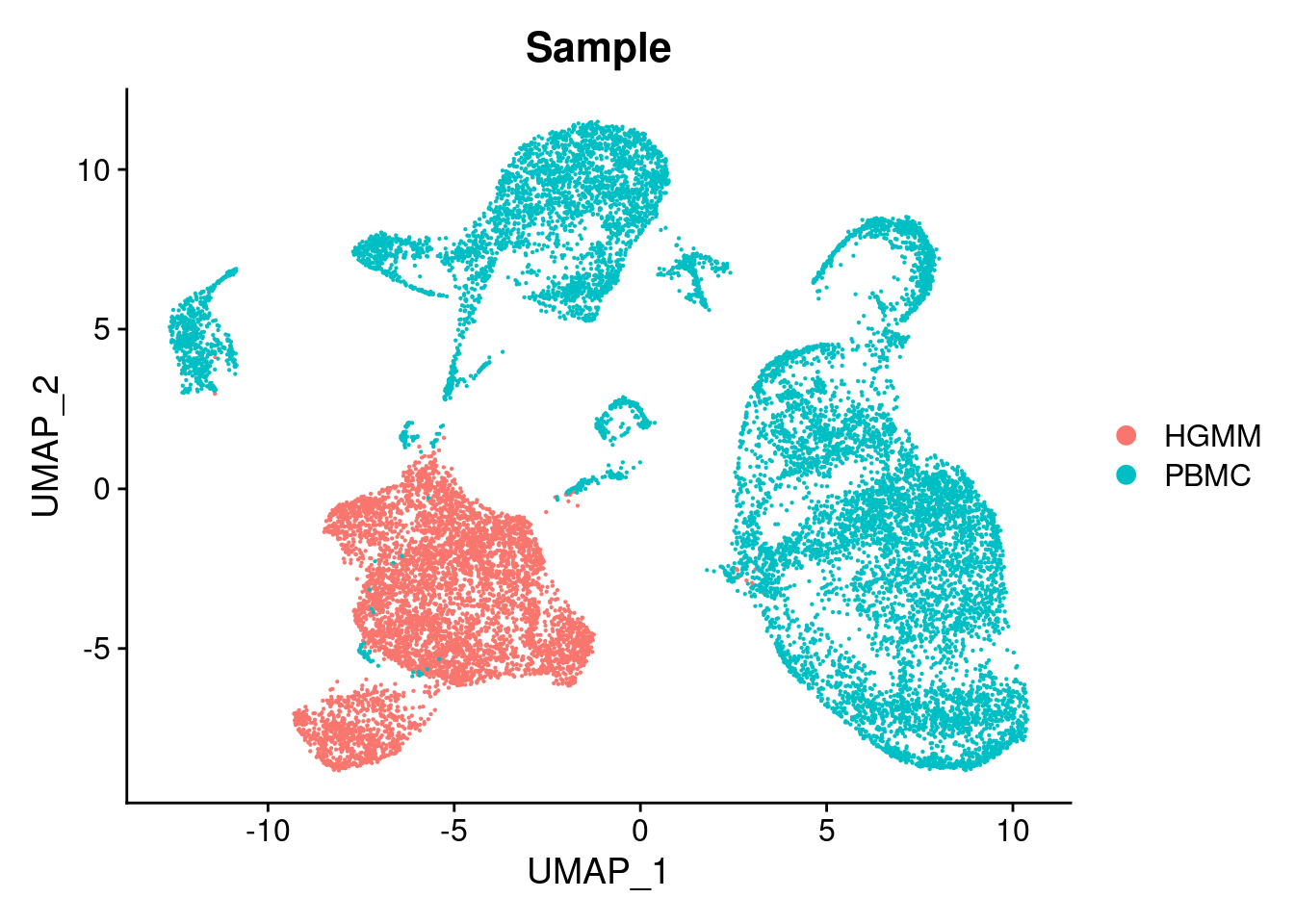
Identify clusters
chromap.integrated <- FindNeighbors(object = chromap.integrated, reduction = 'integrated_lsi', dims = 2:30)Computing nearest neighbor graphComputing SNNchromap.integrated <- FindClusters(object = chromap.integrated, verbose = FALSE, algorithm = 3, resolution = 0.5)
DimPlot(object = chromap.integrated, label = TRUE) + NoLegend()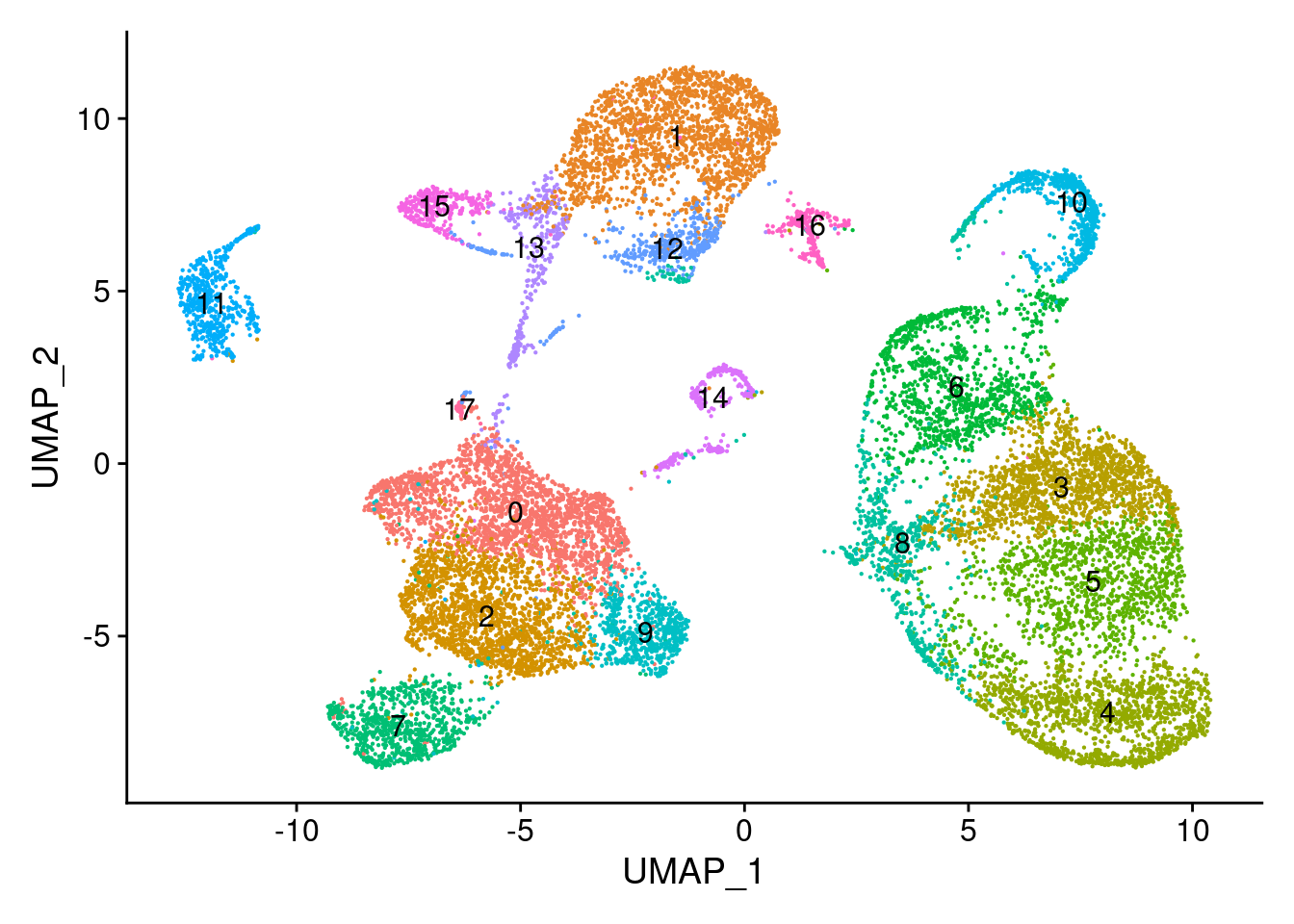
Compute gene activity matrix and insert this as a pseudo RNA assay
gene.activities <- GeneActivity(chromap.integrated)Extracting gene coordinatesExtracting reads overlapping genomic regions
Extracting reads overlapping genomic regions# Add the gene activity matrix to the Seurat object as a new assay and normalize it
chromap.integrated[['RNA']] <- CreateAssayObject(counts = gene.activities)
chromap.integrated <- NormalizeData(
object = chromap.integrated,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(chromap.integrated$nCount_RNA)
)
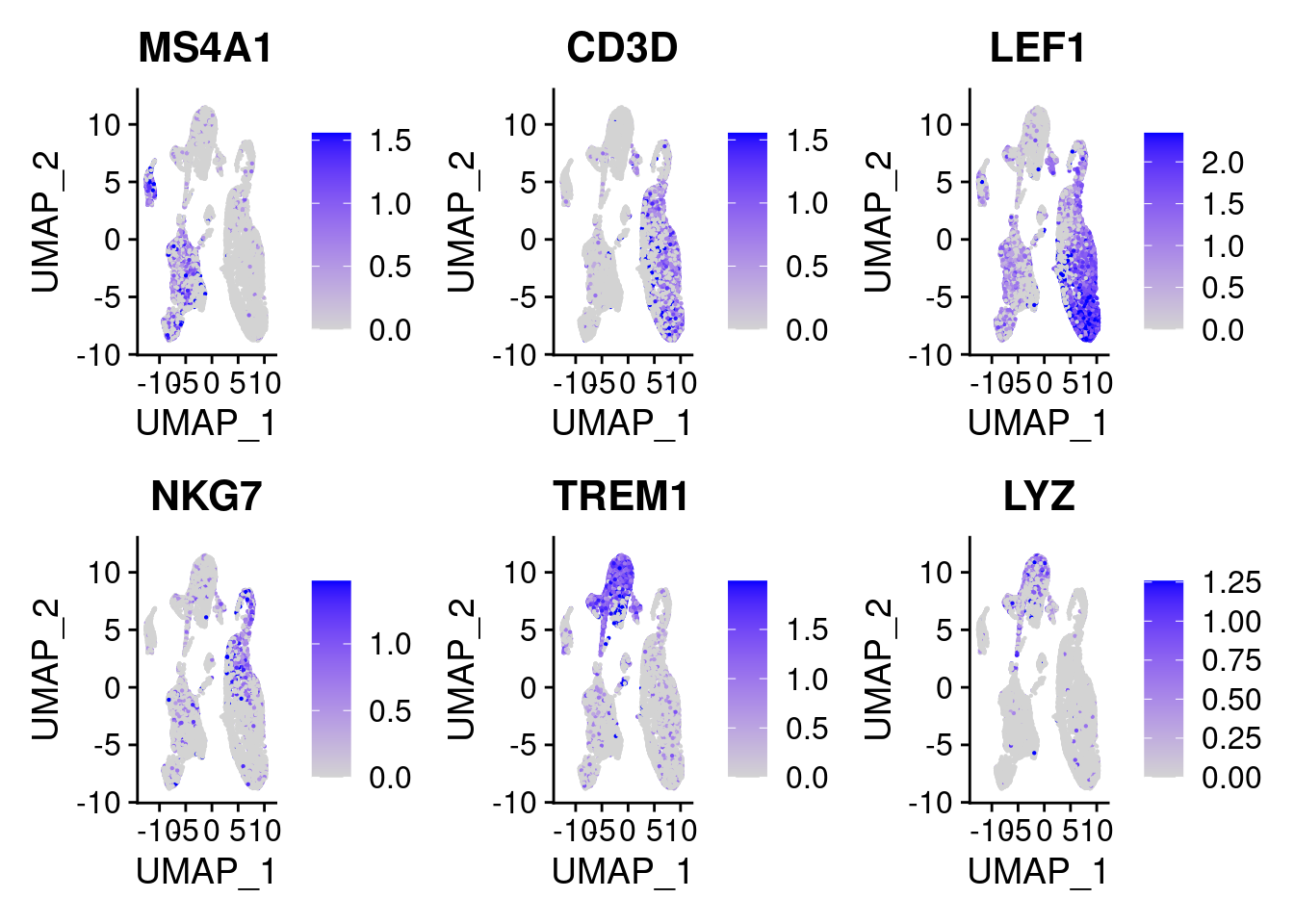
DefaultAssay(chromap.integrated) <- 'RNA'Plot the same marker genes from Seurat vignette
FeaturePlot(
object = chromap.integrated,
features = c('MS4A1', 'CD3D', 'LEF1', 'NKG7', 'TREM1', 'LYZ'),
pt.size = 0.1,
max.cutoff = 'q95',
ncol = 3
)
Save workspace
save.image("cCRE_hg38_10x_HGMM_PBMC_chromap_fragments_MACS_q01_unionPeaks_merge100_seurat_090222_QC_integrated.RData")
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux 8.5 (Ootpa)
Matrix products: default
BLAS/LAPACK: /nas/longleaf/rhel8/apps/r/4.1.0/lib/libopenblas_haswellp-r0.3.5.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] EnsDb.Hsapiens.v86_2.99.0 ensembldb_2.18.3
[3] AnnotationFilter_1.18.0 GenomicFeatures_1.46.5
[5] AnnotationDbi_1.56.2 Biobase_2.54.0
[7] Signac_1.7.0.9003 SeuratObject_4.0.4
[9] Seurat_4.1.0 GenomicRanges_1.46.1
[11] GenomeInfoDb_1.30.1 IRanges_2.28.0
[13] S4Vectors_0.32.4 BiocGenerics_0.40.0
[15] forcats_0.5.1 stringr_1.4.0
[17] dplyr_1.0.9 purrr_0.3.4
[19] readr_2.1.2 tidyr_1.2.0
[21] tibble_3.1.8 ggplot2_3.3.6
[23] tidyverse_1.3.1 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] utf8_1.2.2 reticulate_1.25
[3] tidyselect_1.1.2 RSQLite_2.2.10
[5] htmlwidgets_1.5.4 grid_4.1.0
[7] BiocParallel_1.28.3 Rtsne_0.15
[9] munsell_0.5.0 codetools_0.2-18
[11] ica_1.0-2 future_1.24.0
[13] miniUI_0.1.1.1 withr_2.5.0
[15] spatstat.random_2.1-0 colorspace_2.0-3
[17] filelock_1.0.2 highr_0.9
[19] knitr_1.37 rstudioapi_0.13
[21] ROCR_1.0-11 tensor_1.5
[23] listenv_0.8.0 labeling_0.4.2
[25] MatrixGenerics_1.6.0 git2r_0.30.1
[27] GenomeInfoDbData_1.2.7 polyclip_1.10-0
[29] farver_2.1.0 bit64_4.0.5
[31] rprojroot_2.0.2 parallelly_1.30.0
[33] vctrs_0.4.1 generics_0.1.2
[35] xfun_0.30 BiocFileCache_2.2.1
[37] R6_2.5.1 DelayedArray_0.20.0
[39] bitops_1.0-7 spatstat.utils_2.3-0
[41] cachem_1.0.6 assertthat_0.2.1
[43] BiocIO_1.4.0 promises_1.2.0.1
[45] scales_1.2.0 gtable_0.3.0
[47] globals_0.14.0 processx_3.5.2
[49] goftest_1.2-3 rlang_1.0.4
[51] RcppRoll_0.3.0 splines_4.1.0
[53] rtracklayer_1.54.0 lazyeval_0.2.2
[55] spatstat.geom_2.3-2 broom_1.0.0
[57] yaml_2.3.5 reshape2_1.4.4
[59] abind_1.4-5 modelr_0.1.8
[61] backports_1.4.1 httpuv_1.6.5
[63] tools_4.1.0 ellipsis_0.3.2
[65] spatstat.core_2.4-0 jquerylib_0.1.4
[67] RColorBrewer_1.1-3 ggridges_0.5.3
[69] Rcpp_1.0.8.3 plyr_1.8.7
[71] progress_1.2.2 zlibbioc_1.40.0
[73] RCurl_1.98-1.6 prettyunits_1.1.1
[75] ps_1.6.0 rpart_4.1.16
[77] deldir_1.0-6 pbapply_1.5-0
[79] cowplot_1.1.1 zoo_1.8-9
[81] SummarizedExperiment_1.24.0 haven_2.4.3
[83] ggrepel_0.9.1 cluster_2.1.2
[85] fs_1.5.2 magrittr_2.0.2
[87] RSpectra_0.16-0 data.table_1.14.2
[89] scattermore_0.8 lmtest_0.9-40
[91] reprex_2.0.1 RANN_2.6.1
[93] whisker_0.4 ProtGenerics_1.26.0
[95] fitdistrplus_1.1-6 matrixStats_0.62.0
[97] hms_1.1.1 patchwork_1.1.1
[99] mime_0.12 evaluate_0.15
[101] xtable_1.8-4 XML_3.99-0.9
[103] readxl_1.3.1 gridExtra_2.3
[105] biomaRt_2.50.3 compiler_4.1.0
[107] KernSmooth_2.23-20 crayon_1.5.1
[109] htmltools_0.5.2 mgcv_1.8-40
[111] later_1.3.0 tzdb_0.2.0
[113] lubridate_1.8.0 DBI_1.1.2
[115] dbplyr_2.1.1 rappdirs_0.3.3
[117] MASS_7.3-55 Matrix_1.4-0
[119] cli_3.3.0 parallel_4.1.0
[121] igraph_1.3.3 pkgconfig_2.0.3
[123] GenomicAlignments_1.30.0 getPass_0.2-2
[125] plotly_4.10.0 spatstat.sparse_2.1-0
[127] xml2_1.3.3 bslib_0.3.1
[129] XVector_0.34.0 rvest_1.0.2
[131] callr_3.7.0 digest_0.6.29
[133] sctransform_0.3.3 RcppAnnoy_0.0.19
[135] spatstat.data_2.1-2 Biostrings_2.62.0
[137] rmarkdown_2.12 cellranger_1.1.0
[139] leiden_0.3.9 fastmatch_1.1-3
[141] uwot_0.1.11 restfulr_0.0.13
[143] curl_4.3.2 shiny_1.7.1
[145] Rsamtools_2.10.0 rjson_0.2.21
[147] lifecycle_1.0.1 nlme_3.1-155
[149] jsonlite_1.8.0 viridisLite_0.4.0
[151] fansi_1.0.3 pillar_1.7.0
[153] lattice_0.20-45 KEGGREST_1.34.0
[155] fastmap_1.1.0 httr_1.4.2
[157] survival_3.2-13 glue_1.6.2
[159] png_0.1-7 bit_4.0.4
[161] stringi_1.7.6 sass_0.4.0
[163] blob_1.2.2 memoise_2.0.1
[165] irlba_2.3.5 future.apply_1.8.1